Thalassemia
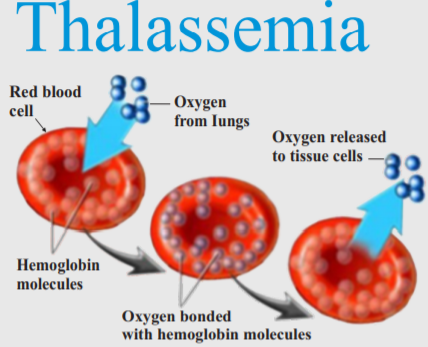
Thalassemia is an inherited blood disorder in which the body makes an abnormal form of hemoglobin. Hemoglobin is the protein molecule in red blood cells that carries oxygen. The disorder results in excessive destruction of red blood cells, which leads to anemia. Anemia is a condition in which your body doesn’t have enough normal, healthy red blood cells.
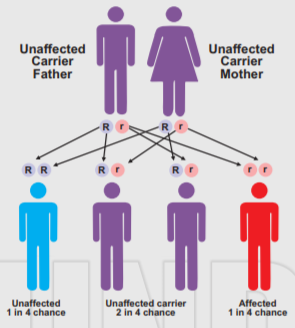
Thalassemia is inherited, meaning that at least one of your parents must be a carrier of the disorder. It’s caused by either a genetic mutation or a deletion of certain key gene fragments. Thalassemia minor is a less serious form of the disorder. There are two main forms of thalassemia that are more serious. In alpha thalassemia, at least one of the alpha globin genes has a mutation or abnormality. In beta thalassemia, the beta globin genes are affected.
Normal Blood
Thalassemia
Each of these forms of thalassemia has different subtypes. The exact form you have will affect the severity of your symptoms and your outlook.
Classifications
At present, thalassemia diseases are classified into transfusion-dependent thalassemia and non-transfusion-dependent thalassemia. This classification is based on the clinical severity of patients determining whether they do require regular blood transfusions to survive (transfusion-dependent thalassemia) or not (non-transfusion-dependent thalassemia). In addition to the previous terminology of “thalassemia major” or “thalassemia intermedia,” this classification has embraced all other forms of thalassemia syndromes such as α-thalassemia, hemoglobin E/β-thalassemia and combined α- and β-thalassemias. Definitive diagnosis of thalassemia and hemoglobinopathies requires a comprehensive workup from complete blood count, hemoglobin analysis, and molecular studies to identify mutations of globin genes.
Signs and Symptoms
There are several types of thalassemia. The signs and symptoms you have depend on the type and severity of your condition. Thalassemia signs and symptoms can include:
- Fatigue
- Weakness
- Pale or yellowish skin
- Facial bone deformities
- Slow growth
- Abdominal swelling
- Dark urine
Some babies show signs and symptoms of thalassemia at birth; others develop them during the first two years of life. Some people who have only one affected hemoglobin gene don't have thalassemia symptoms.
Causes
Thalassemia is caused by mutations in the DNA of cells that make hemoglobin — the substance in red blood cells that carries oxygen throughout your body. The mutations associated with thalassemia are passed from parents to children. Hemoglobin molecules are made of chains called alpha and beta chains that can be affected by mutations. In thalassemia, the production of either the alpha or beta chains are reduced, resulting in either alpha-thalassemia or beta-thalassemia.
In alpha-thalassemia, the severity of thalassemia you have depends on the number of gene mutations you inherit from your parents. The more mutated genes, the more severe your thalassemia. In beta-thalassemia, the severity of thalassemia you have depends on which part of the hemoglobin molecule is affected.
Diagnosis
Most children with moderate to severe thalassemia show signs and symptoms within their first two years of life. If your doctor suspects your child has thalassemia, he or she can confirm a diagnosis with blood tests. Blood tests can reveal the number of red blood cells and abnormalities in size, shape or color. Blood tests can also be used for DNA analysis to look for mutated genes.
Treatments
Mild forms of thalassemia trait don't need treatment. For moderate to severe thalassemia, treatments might include:
- Frequent blood transfusions. More severe forms of thalassemia often require frequent blood transfusions, possibly every few weeks. Over time, blood transfusions cause a buildup of iron in your blood, which can damage your heart, liver and other organs.
- Chelation therapy. This is treatment to remove excess iron from your blood. Iron can build up as a result of regular transfusions. Some people with thalassemia who don't have regular transfusions can also develop excess iron. Removing the excess iron is vital for your health.
- To help rid your body of the extra iron, you might need to take an oral medication, such as deferasirox (Exjade, Jadenu) or deferiprone (Ferriprox). Another drug, deferoxamine (Desferal), is given by needle.
- Stem cell transplant. Also called a bone marrow transplant, a stem cell transplant might be an option in some cases. For children with severe thalassemia, it can eliminate the need for lifelong blood transfusions and drugs to control iron overload. This procedure involves receiving infusions of stem cells from a compatible donor, usually a sibling.
Complications
The life of patients with thalassemia has improved both in duration and in quality in industrialized countries. Complications are still common and include heart disease (heart failure and arrhythmias), chronic liver hepatitis, which can evolve in cirrhosis and, rarely, in hepatocellular carcinoma, endocrine problems (hypogonadism, hypothyroidism, diabetes, hypoparathyroidism), stunted growth, osteoporosis, thrombophilia and pseudoxanthoma elasticum.
The incidence of complications is decreasing in younger cohorts of patients who have been transfused with blood that has been screened for viruses and thanks to the introduction of new oral iron chelators and imaging methods. The accurate measurement of iron deposits allows better management of iron overload. In addition, therapy for several complications is available. Specialized competence in treating patients with thalassemia is of great importance.
Epidemiology
Thalassemia is the most common monogenic disorder worldwide. It is common in areas with prevalent malaria as thalassemic red cells provide immunity against the parasite. The incidence of Thalassemia carriers is high in regions such as Mediterranean, Middle East, Indian subcontinent, Southeast Asia and South China. In the past few decades, migrants from the Thalassemia prevalent countries to non-prevalent countries, mainly North America and Central and North Europe, are rapidly increasing in number.
The non-prevalent countries may not have established pre-natal screening system for Thalassemia. The genetic subtypes among the different ethnic groups vary; this may pose challenges in prenatal diagnosis. Genetic counselling on the postnatal course of Thalassemia may be affected by the genotype-phenotype correlation and coinheritance of other genetic diseases. New treatment methods improve the survival of patient with Thalassemia major, but some late complications that occur with longer survival have been recently discovered.