Hemophilia
Hemophilia is a rare disorder in which your blood doesn't clot normally because it lacks sufficient blood-clotting proteins (clotting factors). If you have hemophilia, you may bleed for a longer time after an injury than you would if your blood clotted normally.
The major types of this condition are hemophilia A (also known as classic hemophilia or factor VIII deficiency) and hemophilia B (also known as Christmas disease or factor IX deficiency). Although the two types have very similar signs and symptoms, they are caused by mutations in different genes. People with an unusual form of hemophilia B, known as hemophilia B Leyden, experience episodes of excessive bleeding in childhood but have few bleeding problems after puberty.
SIGNS & SYMPTOMS
Signs and symptoms of hemophilia vary, depending on your level of clotting factors. If your clotting-factor level is mildly reduced, you may bleed only after surgery or trauma. If your deficiency is severe, you may experience spontaneous bleeding. Signs and symptoms of spontaneous bleeding include:
- Unexplained and excessive bleeding from cuts or injuries, or after surgery or dental procedures
- Many large or deep bruises
- Unusual bleeding after vaccinations
- Pain, swelling or tightness in your joints
- Blood in your urine or stool
- Nosebleeds without a known cause
- In infants, unexplained irritability
Complications
Complications of hemophilia may include:
- Deep internal bleeding. Bleeding that occurs in deep muscle can cause your limbs to swell. The swelling may press on nerves and lead to numbness or pain.
- Damage to joints. Internal bleeding may also put pressure on your joints, causing severe pain. Left untreated, frequent internal bleeding may cause arthritis or destruction of the joint.
- Adverse reaction to clotting factor treatment. In some people with severe hemophilia, the immune system has a negative reaction to the clotting factors used to treat bleeding. When this happens, the immune system develops proteins (known as inhibitors) that inactivate the clotting factors, making treatment less effective.
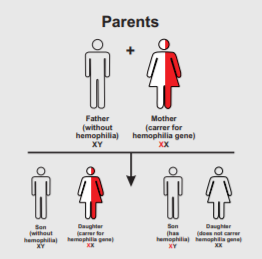
Genetics
Typically, females possess two X-chromosomes, and males have one X and one Y-chromosome. Since the mutations causing the disease are X-linked recessive. A female carrying the defect on one of her X-chromosomes may not be affected by it, as the equivalent dominant allele on her other chromosome should express itself to produce the necessary clotting factors, due to X inactivation. Therefore, heterozygous females are just carriers of this genetic disposition. However, the Y-chromosome in the male has no gene for factors VIII or IX. If the genes responsible for production of factor VIII or factor IX present on a male's X-chromosome are deficient there is no equivalent on the Y-chromosome to cancel it out, so the deficient gene is not masked and the disorder will develop.
A mother who is a carrier, has a 50% chance of passing the faulty X-chromosome to her daughter, while an affected father will always pass on the affected gene to his daughters. A son cannot inherit the defective gene from his father. If a female gives birth to a hemophiliac son, either the female is a carrier for the blood disorder or the hemophilia was the result of a spontaneous mutation. If a male is afflicted with the disease and has children with a female who is not a carrier, his daughters will be carriers of hemophilia. His sons, however, will not be affected with the disease. The disease is X-linked and the father cannot pass hemophilia through the Y-chromosome. Males with the disorder are then no more likely to pass on the gene to their children than carrier females, though all daughters they sire will be carriers and all sons they father will not have hemophilia (unless the mother is a carrier).
Severity
There are numerous different mutations which cause each type of hemophilia. Due to differences in changes to the genes involved, people with hemophilia often have some level of active clotting factor. Individuals with less than 1% active factor are classified as having severe hemophilia, those with 1-5% active factor have moderate hemophilia, and those with mild hemophilia have between 5-40% of normal levels of active clotting factor.
Diagnosis
Hemophilia can be diagnosed before, during or after birth if there is a family history of the condition. Several options are available to parents. If there is no family history of hemophilia, it is usually only diagnosed when a child begins to walk or crawl. They may experience joint bleeds or easy bruising. Mild hemophilia may only be discovered later, usually after an injury or a dental or surgical procedure.

Clotting Factor
Clotting factors are usually not needed in mild hemophilia. In moderate hemophilia clotting factors are typically only needed when bleeding occurs or to prevent bleeding with certain events. In severe hemophilia preventive use is often recommended two or three times a week and may continue for life. Rapid treatment of bleeding episodes decreases damage to the body.
Prognosis
Like most aspects of the disorder, life expectancy varies with severity and adequate treatment. People with severe hemophilia who don't receive adequate, modern treatment have greatly shortened lifespans and often do not reach maturity. Prior to the 1960s when effective treatment became available, average life expectancy was only 11 years. By the 1980s the life span of the average hemophiliac receiving appropriate treatment was 50–60 years. Today with appropriate treatment, males with hemophilia typically have a near normal quality of life with an average lifespan approximately 10 years shorter than an unaffected male.

